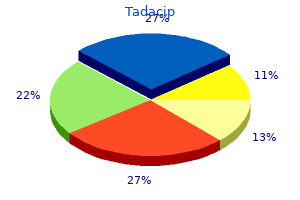
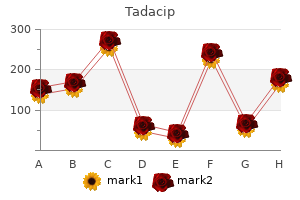
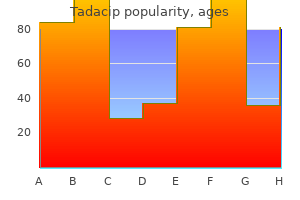
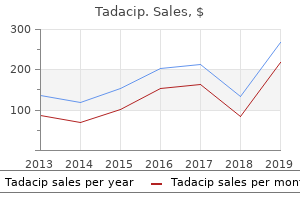
Tadacip
"Order tadacip 20mg with amex, erectile dysfunction homeopathic treatment".
By: Y. Grompel, M.B. B.CH. B.A.O., Ph.D.
Assistant Professor, University of Nevada, Reno School of Medicine
Rarely impotence remedy order genuine tadacip on line, the lesions appear as a depressed area of atrophy ("atrophic variant")8 or as subcutaneous masses without obvious connection to the overlying dermis erectile dysfunction treatment lloyds generic tadacip 20mg with amex. Less often, multiple small subcutaneous nodules appear initially rather than a plaque. The plaque may grow slowly or remain stationary for a variable period, eventually entering a more rapid growth phase and giving rise to one or more nodules. Thus, only in the fully developed lesion is the typical "protuberant" appearance manifested. Neglected tumors may achieve enormous proportions and have multiple satellite nodules. Despite the large size of many of these tumors, patients appear surprisingly well and lack the signs of cachexia associated with malignancies. Over the years it has been considered a fibroblastic, histiocytic, and neural tumor. Rarely, dermatofibrosarcomas are centered in the subcutis, with only subtle dermal involvement. Hemorrhage and cystic change are sometimes seen, but necrosis, a common feature of undifferentiated pleomorphic sarcomas, is rare. The tumor may reach the epidermis or leave an uninvolved zone of dermis just underneath the epidermis. In either event, the overlying epidermis does not usually display the hyperplasia that characterizes some cutaneous fibrous histiocytomas (dermatofibromas). Spread of the tumor between preexisting collagen of the dermis may simulate the appearance of a cutaneous fibrous histiocytoma (A). There is usually little nuclear pleomorphism and only low to moderate mitotic activity. Secondary elements such as giant cells, xanthoma cells, and inflammatory elements are few in number or absent altogether. These myxoid areas occur in both primary and recurrent lesions and are characterized by the interstitial accumulation of ground substance material. As myxoid change of the stroma becomes more pronounced, the storiform pattern becomes less distinct and the vascular pattern more apparent. Confident diagnosis of highly myxoid dermatofibrosarcomas usually requires identification of more typical areas. Giant cells, similar to those in giant cell fibroblastoma, can be identified in a small percentage of otherwise typical dermatofibrosarcomas. Originally construed as evidence of myofibroblastic differentiation,10 these structures seem to be centered in some cases around blood vessels,11,12 and likely represent an unusual nonneoplastic vascular response to the tumor. Characterized by long fascicles of spindle cells with more nuclear atypia and mitotic activity, these areas usually sharply abut conventional low-grade areas. B, When myxoid change is prominent, storiform pattern may be lacking altogether, and tumor may resemble a myxoid liposarcoma. Fibrosarcomatous areas were originally believed to be more common in recurrent lesions, but studies have documented that the contrary is true. Apolipoprotein D may also be of value in the distinction of dermatofibrosarcomas from fibrous histiocytomas, although this antibody is used in relatively few laboratories. The distinction between benign fibrous histiocytoma and dermatofibrosarcoma occasionally proves difficult when only the superficial portion of the dermatofibrosarcoma is present in a biopsy specimen, because these areas appear so well differentiated (Table 11. Under these circumstances, knowledge of the size and configuration of the lesion in question suggests the diagnosis, and biopsy of a deeper portion confirms it. Moreover, its typical deep location in muscle and more rapid growth are at variance with the indolent course of this tumor. As indicated earlier, when such areas represent more than just a microscopic focus, they should be diagnosed as "sarcoma arising in dermatofibrosarcoma protuberans. This is most likely to occur when dermatofibrosarcoma is in the plaque stage or when a biopsy is done on only the periphery of the tumor. However, neurofibroma usually contains tactoid structures or other features of neural differentiation, and it lacks the highly cellular areas with mitotic figures that characterize the central portion of a dermatofibrosarcoma. Highly myxoid forms of dermatofibrosarcoma may resemble myxoid liposarcoma by virtue of the prominent vasculature and bland stellate or fusiform cells.
The peripheral areas may also resemble neurofibroma impotence purchase generic tadacip online, but the myofibroblastic cells lack S-100 protein erectile dysfunction cleveland clinic quality 20 mg tadacip. Fibrous histiocytoma is composed of a polymorphous proliferation of cells arranged in a more pronounced storiform pattern. The latter tends to be less well circumscribed, arise in muscle, and show a more uniform spindle cell pattern. In addition, lipofibromatosis shows neither central necrosis nor a central hemangiopericytoma-like vascular pattern. Such lesions include Ewing sarcoma, mesenchymal chondrosarcoma, malignant solitary fibrous tumor, and poorly differentiated synovial sarcoma. Although not always present, identifying peripheral myoid-appearing cells is the most useful feature for recognizing myofibromatosis. Sinonasal glomangiopericytoma is unique to the nasal cavity and passages and has no counterpart in soft tissue proper. The majority are polypoid lesions that involve the nasal cavity or paranasal sinuses and grow as diffuse submucosal masses encircling minor salivary glands. Spindled to oval cells are arranged in short fascicular, storiform, whorled, or mixed patterns. The cells within sinonasal glomangiopericytoma have a distinctly myoid phenotype despite that they do not resemble mature smooth muscle cells. Solitary and multiple lesions confined to soft tissues and bone (with no evidence of visceral involvement) carry an excellent prognosis; they tend to regress spontaneously and rarely require more than a diagnostic biopsy. In addition, 11 of 18 patients (61%) with multicentric lesions without visceral involvement and follow-up of more than 1 year had spontaneous regression of the lesions. Chung and Enzinger100 found that only 3 of 28 solitary lesions (11%) locally recurred, and several of the multicentric lesions without visceral involvement showed spontaneous regression. The natural history of highly cellular, atypical variants of myofibroma appears to be identical to that of their more conventional counterparts. As many as 75% die with signs of respiratory distress or diarrhea soon after birth,100,101,132 although there are exceptions. A, the rounded to spindled myoid cells are arranged around an intricate vasculature. These consist of missense mutations, with amino acid substitutions clustering at positions 33 to 45, and corresponding to the recognition site of the -catenin destruction complex. Patients at greatest risk to die of their disease are those with a long history of symptoms or those whose tumors display marked atypia or bone invasion. A case of multiple subungual glomus tumors associated with neurofibromatosis type 1. Gastrointestinal glomus tumors: a clinicopathologic, immunohistochemical, and molecular genetic study of 32 cases. Hereditary multiple glomus tumors involving the glans penis: a case report and review of the literature. High-flow priapism due to a malignant glomus tumor (glomangiosarcoma) of the corpus cavernosum. Atypical glomus tumor in the mediastinum: a case report with immunohistochemical and ultrastructural studies. Pulmonary and mediastinal glomus tumors-report of five cases including a pulmonary glomangiosarcoma: a clinicopathologic study with literature review. Immunohistochemistry in the differential diagnosis of nodular hidradenoma and glomus tumor. Cutaneous glomus tumor: a comparative immunohistochemical study with pseudoangiomatous intradermal melanocytic nevi. The immunophenotype of hemangiopericytomas and glomus tumors, with special reference to muscle protein expression: an immunohistochemical study and review of the literature. Notch2 and Notch3 function together to regulate vascular smooth muscle development. Locally infiltrative glomus tumors and glomangiosarcomas: a clinical, ultrastructural, and immunohistochemical study. Angiomatosis of soft tissue: an analysis of the histologic features and clinical outcome in 51 cases.
Primary cutaneous osteosarcoma of the skin: a report of 2 cases with emphasis on the differential diagnoses erectile dysfunction causes divorce generic 20mg tadacip free shipping. Postradiation extraskeletal osteosarcoma masquerading as an axillary artery pseudoaneurysm erectile dysfunction doctors naples fl discount 20mg tadacip overnight delivery. Primary osteogenic sarcoma of the breast: a clinicopathologic analysis of 50 cases. Evidence indicates that the cells in these lesions are fibroblastic or have some features of myofibroblasts. The disorder occurs predominantly in otherwise healthy children, adolescents, and young adults; is more often multiple than solitary; and can affect two or more siblings of the same family. Unlike similar calcifications associated with renal insufficiency, hypervitaminosis D, and milk-alkali syndrome, there are no demonstrable abnormalities in calcium metabolism. The term tumoral calcinosis was coined by Inclan1 in 1943, but this condition was recognized as an entity much earlier. In 1899, Duret2 observed this process in siblings, a 17-year-old girl and her younger brother, who had multiple calcifications in the vicinity of the hip and elbow joint. Later, in 1935, Teutschlaender3 gave a detailed account of another typical case, an 11-year-old girl with multiple lesions in the shoulder and elbow regions that had onset at age 2 years. He thought this process was secondary to fat necrosis and used the term lipid calcinosis. Since these descriptions, numerous other acceptable examples of this growth have been reported under various names, including calcifying bursitis,4 calcareous tendinitis,5 and "Kikuyu bursa. Most common is the sporadic (nonfamilial) and idiopathic form, with onset during the first and second decades of life and rare in patients older than 50. It affects whites and blacks about equally, and there is a slight female preponderance. Most patients present with a solitary large, firm, subcutaneous calcified mass that is slowly growing and usually asymptomatic, typically located in the vicinity of a large joint,1 especially the trochanteric and gluteal regions of the hip and the lateral portion of the shoulder and the posterior elbow (Table 31. The lesion is firmly attached to the underlying fascia, muscle, or tendon and may even infiltrate these structures, but it is unrelated to bone, and the underlying joints are unaffected. The familial form of tumoral calcinosis has two variants, hyperphosphatemic and normophosphatemic, both inherited in an autosomal recessive manner but characterized by distinct genetic mutations. These patients characteristically have elevation of serum phosphate and vitamin D, unless they have the normophosphatemic variant. The lesions are often multifocal and may be associated with a number of other bony abnormalities, including calcifications in the shaft of long bones and cranium as well as ocular and dental abnormalities. Despite the large amounts of calcium in these lesions in patients with idiopathic tumoral calcinosis, there is no evidence of osteoporosis in the skeleton, as seen in patients with renal insufficiency and secondary hyperparathyroidism. In the active (cellular) phase, a central mass of amorphous or granular calcified material is bordered by a florid proliferation of mononucleated or multinucleated macrophages, osteoclast-like giant cells, fibroblasts, and chronic inflammatory elements. During the inactive phase, there is merely calcified material surrounded by dense fibrous material extending into the adjacent tissues, or a cystic space surrounded by calcium deposits. Differential Diagnosis Morphologically, the lesions of tumoral calcinosis are identical regardless of whether they are idiopathic, familial, or secondary. Patients with chronic renal disease and secondary hyperparathyroidism are usually older than those with idiopathic tumoral calcinosis, have additional calcifications in visceral organs. Nine months after calcified mass in the right hip (A) was removed, a second mass developed in the left hip (B). Patients with excessive osteolysis and mobilization of calcium in destructive neoplastic and infectious process of bone may also develop lesions that can resemble tumoral calcinosis. In all these lesions, a detailed clinical history and laboratory data aid in reaching a reliable diagnosis. Some of these cases could represent "tenosynovitis with psammomatous calcifications. Histologically, the lesion shows tendinous degeneration associated with psammomatous calcifications and a histiocyte-rich infiltrate. Calcinosis cutis universalis and calcinosis cutis circumscripta likewise are located in the skin and subcutis and are associated with normal serum calcium and phosphorus levels. Calcinosis universalis forms multiple nodules or plaques mainly in children and in about half the cases associated with manifestations of scleroderma, systemic lupus erythematosus, or dermatomyositis. Calcinosis circumscripta, on the other hand, chiefly affects middle-aged women and typically involves the hand and wrist, including tendon sheaths.

No matter how selective the antibodies or how powerful the detection system erectile dysfunction quiz buy tadacip master card, the method fails if the analytic tools are inadequate erectile dysfunction in diabetes mellitus pdf 20 mg tadacip free shipping. The expression of certain antigens, or clusters of antigens, is characteristic of some tumors. Whereas thousands of monoclonal and polyclonal antibodies are available to assist in tumor diagnosis, only a small subset has proved to be of practical value in the diagnosis of soft tissue neoplasms. The original thinking that intermediate filament expression was restricted to specific cell types. The following sections on intermediate filaments concentrate not only on the normal pattern of expression of these proteins, but also on the situations where intermediate filaments show "anomalous expression. Vimentin is ubiquitously expressed in all cells during early embryogenesis and is gradually replaced in many cells by type-specific intermediate filaments. Vimentin is expressed in virtually all mesenchymal tumors and is thus of minimal value in identifying particular tumors. Given the frequent coexpression of vimentin along with keratin in carcinomas, vimentin expression is also of little value in the immunohistochemical distinction of carcinomas from sarcomas. Schwann cell Adipocyte Chondrocyte Osteocyte Myofibroblast Interstitial cells of Cajal Classification of human epithelium and their neoplasms using monoclonal antibodies to keratins: strategies, applications, and limitations. However, vimentin expression, similar to that of all the intermediate filaments, is rather hardy and may remain present in tissues in which all other immunoreactivity has been lost. Keratins Keratins, also known as cytokeratins, the most complex members of the intermediate filament protein family, are a collection of more than 20 proteins. Keratins are highly sensitive markers for identifying carcinomas and are generally employed as markers distinguishing epithelial/mesothelial from nonepithelial tumors. Over the past decade it has become clear that keratin expression is not restricted to carcinomas. Anomalous keratin expression is typically characterized by immunostaining (even under optimal technical conditions) in only a subset of the target cell population. In these cells, keratin is present in only a portion of the cytoplasm, often yielding a "perinuclear" or "dotlike" pattern of immunostaining. This dotlike pattern is not always an indication of anomalous keratin, however, because it is typically seen in some neuroendocrine carcinomas. It is instead a feature of a limited subset of nonepithelial tumors, particularly smooth muscle tumors, melanomas, and endothelial cell tumors; as such, it may serve as a clue to the diagnosis of these tumors. Subsequent studies have shown that at least 30% of leiomyosarcomas manifest keratin. Demonstration of strong keratin expression is useful for distinguishing this entity from cystic lymphangioma. Despite that many studies completed during the mid-1980s concluded that melanomas were vimentin-positive, keratin-negative tumors,29 Zarbo et al. They also performed one- and two-dimensional gel electrophoresis with immunoblotting, confirming that keratin 8 was expressed by the tumor cell population. Although anomalous keratin expression was previously thought to be more common in metastatic than in primary melanomas,32 this does not seem to be the case. Early reports suggested that vascular tumors manifesting epithelioid histologic features. The largest published series of angiosarcomas of deep soft tissue documented keratin expression in about one-third of cases. A surprising number of tumors in the category of "small, blue round cell tumors" of childhood typically coexpress keratin in a pattern similar to that of anomalous keratin expression. These tumors include Ewing sarcoma,41-44 rhabdomyosarcoma,45,46 Wilms tumor,47,48 and desmoplastic small round cell tumor of childhood. Poorly differentiated synovial sarcoma (B) demonstrating focal expression of high-molecular-weight keratins. C, Expression of keratin in synovial sarcomas may be focal, and some express only high-molecular-weight isoforms. Dotlike expression of keratin and other intermediate filaments is not specific for neuroendocrine carcinomas and may be seen in any "small, blue round cell. Vascular tumors, particularly epithelioid ones, typically express low-molecular-weight keratins and may be mistaken for carcinoma.


